")
")
Huntingtin: low-complexity regions at high-resolution (PI: Pau Bernadó)
People Involved: P. Bernadó, N. Sibille, F. Allemand, A. Urbanek, A. Morató, A. Fournet, M. Popovic, C. Elena
While most protein sequences are aperiodic and feature most of the 20 amino acids, many proteins harbor low complexity regions (LCRs), with a highly biased composition. LCRs are functionally re- levant and, in some cases, are directly related with severe diseases. Despite their relevance, high-resolution structural and dynamic characterization of LCRs cannot be tackled with current methods placing them on the dark side of proteome.
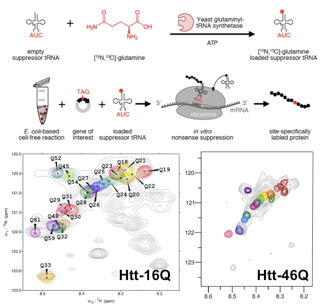
Homorepeats, a subclass of LCRs that is characterized by stretches of the same amino acid, perform very specialized functions facilitated by the localized enrichment of the same physicochemical property. In addition, numerous severe pathologies have been associated with abnormally long repetitions eg. huntingtin (Htt). The N-terminal region of Htt, known as exon-1, contains glutamine and proline stretches, and is the prototypical example of a homorepeat. The poly-Gln tract is directly linked to Huntington's disease (HD), a deadly neuropathy appearing in individuals with more than 35 consecutive Gln residues, the pathological threshold. Present structural biology approaches do not allow high-resolution studies of Htt to investigate the origin of the pathological threshold. Our group is developing chemical biology tools to enable the residue-specific isotopic labeling of Htt. This allows us for first time to study Htt at the atomic level with NMR. The applica- tion of these approaches to several Htt constructs with Q tracts of diffe- rent lenghts will shed light on the structural bases of the pathological threshold of HD.
Main collaborators: S. Delbecq (U. Montpellier), C. Cativiela (Zaragoza, Spain), J. Cortés (LAAS, Toulouse)
Grant: ANR SPIN-HD, ERC ChemRepeat
Molecular mechanisms of functional disordered C-terminal regions of GPCRs and impact on arrestin signaling pathways (PI: Nathalie Sibille)
People Involved: N. Sibille, P. Bernadó, F. Allemand, A. Fournet, A. Thureau, A. Mouhand, M. Guillien
This project aims to elucidate the details and principles of non-G protein dependent signaling. Arrestin-mediated and ligand-induced biased signaling is a hot topic currently in GPCR and drug research in general. By focusing on ghrelin, b2ar and V2 receptors, we have chosen both varied GPCRs to investigate by state-of-the-art biochemical and especially biophysical methods the func- tional disordered regions of GPCRs with regard to their interaction with arrestins.
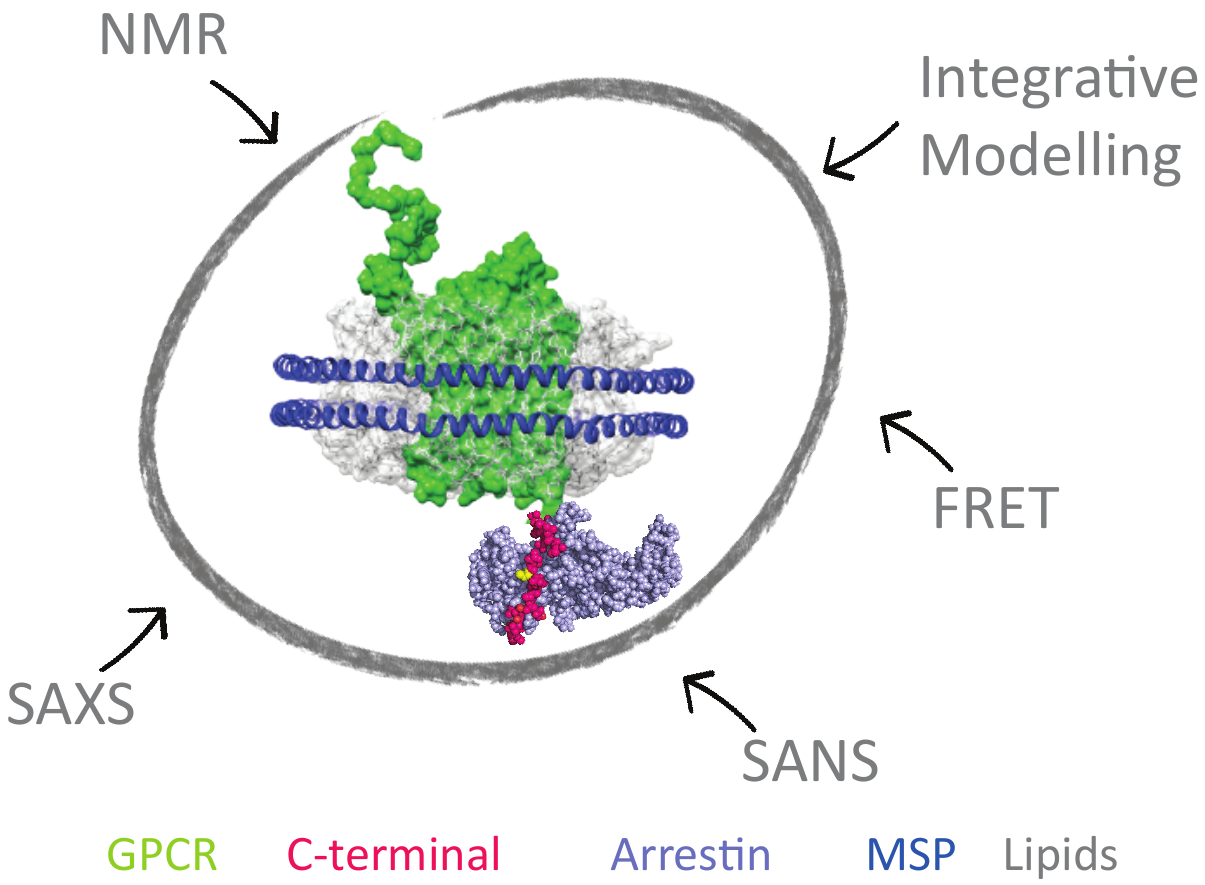
To design more effective drugs without side effects, it is essential to better understand the molecular mechanism of GPCR (G-Protein Coupled Receptors), which are targeted by one third of drugs on the market. Some crucial cell signaling pathways are mediated by the interaction of the cytoplasmic C-ter- minal part of the GPCR with β-arrestin. This functional interaction is modulated by GPCR-associated kinases (GRK). This project aims at revealing the link between C-tail phosphorylation patterns by the various GRKs, their structural dynamics and the different related arrestin «func- tional conformations». This will be achieved by combining solution spectroscopic techniques, such as NMR and SAS, on model systems of increasing complexity ranging from isolated peptides to purified signaling complexes into membrane-mi- micking systems (nanodiscs). This study will reveal the struc- tural and dynamic mechanisms necessary for the interaction of the C-terminal regions of GPCRs with β-arrestin and provide essential information to guide the rational design of peptide mimetics able to modulate specific signaling cascades.
Main collaborators: J.L. Banères (IBMM, Montpellier), B. Mouillac (IGF, Montpellier), L. Arleth (U. Copenhagen)
Grant: ANR GPCteR
Regulation of gene transcription by retinoic acid receptor heterodimer and its coregulators (PIs: N. Sibille & P. Bernadó)
People Involved: N. Sibille, P. Bernadó, F. Allemand, A. Fournet, T. Cordeiro, L. Sénicourt, A. Sagar
Nuclear Receptors (NRs) are transcription factors with a direct role in regulating the expression of hormone-response genes. Agonist binding to NRs triggers transcription through a cascade of events that is initiated by the exchange of the repression complex by the activation one. The assembly of the two large complexes is mediated by intrinsically disordered coregulator proteins that, simultaneously, also interact with the NRs. Coregulators are named coactivators or corepressors when they mediate the activation or repression complexes, respectively. In collaboration with A. le Maire, W. Bourguet and P. Germain we have dissected the structural bases of the interaction between both families of coregulators with the Retinoic Acid Receptor heterodimer (RAR/RXR). Funded by the Labex EpiGenMed, we have studied the interaction of the intrinsically disordered NCoR (corepressor) and TIF2 (coactivator) with RAR/RXR, which form highly dynamic and malleable complexes.
![]()
We reached to the following conclusions for the corepressor NCoR (Cordeiro et al., 2019): (i) Small ligands induce disorder to order transitions in the C-
terminal helix of the Ligand Binding Domain (LBD) of both RAR and RXR, and they occur
independently of the presence of coregulator proteins.
(ii) We demonstrated that the NR binding region of NCoR is intrinsically disordered, but it encompasses three highly conserved regions that are partially structured that are linked to RAR/RXR recognition. (iii) The NCoR complex with RAR/RXR is highly plastic as N-CoR is bound to the heterodimer in a singly- and a doubly-bound form (Fig. 3). The relative populations of these complexes can be modulated with small ligands and point mutations.
The main results derived from the study of the coactivator TIF2 are (Sénicourt et al.,
in preparation): (i) We demonstrated by NMR that the NR binding region of Tif2 is intrinsically disordered, but it encompasses three highly conserved regions that are partially structured. (ii) In addition of the three well-known NR interacting sites, we have highlighted importance of a flanking region in strengthening the interaction with RAR/RXR. Initially identified by NMR, this flanking region has been successfully co-crystalized with the nuclear receptor, displaying a helix-turn-helix motif. Further studies will be performed to dissect the relevance of this interaction.
These results provide the structural and functional clues for the regulation of gene transcription and suggest mechanistic pathways for the transition from basal (inactive) state to the transcriptionally active one.
Main collaborators: A. LeMaire, P. Germain, W. Bourguet, A. Barducci (CBS, Montpellier)
Grant: Labex EpiGenMed
Computational tools for modeling and data analyses (PIs: P. Bernadó & N. Sibille)
People Involved: P. Bernadó, N. Sibille, F. Allemand, F. Herranz-Trillo, A. Estana, A. Sagar
During the present contract the group has developed an intense activity in developing new tools for the structural modeling of flexible systems as well as the comprehensive analysis of complex SAXS data. In the following paragraphs the most relevant results on these two research lines will be described.
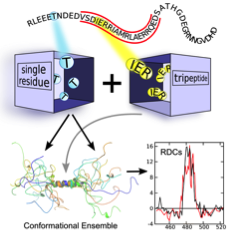
- Structural modeling of Disordered Proteins: Structural modeling of IDPs is extremely challenging as their special features are coded in the conformational bias at the residue-level. In order to solve of these challenges, we have developed a tripeptide database from high-resolution structures in collaboration with Juan Cortés that allows to build realistic IDP ensemble models for these proteins (Estaña et al, 2019). The resulting ensembles are in agreement with extensive experimental NMR and SAXS data. The enhanced performance of this approach is based on the inclusion of the specific chemical and structural properties of the neighboring residues. The tripeptide database is extremely rich and can also be used to decipher the folding pathway of small structural elements of proteins (Estaña et al., 2019) and the prediction of partially structured elements in IDPs from their sequences (Estaña et al., in preparation).
- Chemometric analysis of SAXS data from complex biomolecular systems: Many biological systems present distinct species that are in equilibrium. These polydisperse systems are extremely difficult to characterize as the experimental observables are population-weighted averages. SAXS is very sensitive to the overall size and shape of particles and, therefore, it can monitor changes when the equilibrium is perturbed by modifying the experimental conditions (concentration, pH, time...). We have developed a chemometric approach, which we call COSMiCS, to decompose large population-weighted SAXS datasets. COSMiCS uses multiple SAXS data representations to decrease the degeneracy of possible solutions. As a proof of concept, we applied COSMiCS to study the fibrillation of insulin and α-synuclein by SAXS for which the population and structure of the three main species (monomer, oligomer and fibril) in equilibrium were derived (Herranz-Trillo et al., 2017).
Main collaborators: J. Cortés (LAAS, Toulouse), B. Vestergaard (U. Copenhagen), R. Tauler (CSIC, Bercelona)