Theme 1: Mechanisms of gene regulation by nuclear receptors
P Germain, A le Maire, W Bourguet
The overall goal of this theme is to use a combination of biochemical, structural, cellular, genomic and in vivo approaches to gain mechanistic insights into the general principles governing nuclear receptor action and regulation.
The precise regulation of gene expression requires activation and repression of DNA transcription and both activities appear critical for the biological functions of a number of receptors including the retinoic acid receptors (RARα, β, γ) that form functional heterodimers with retinoid X receptors (RXRα, β, γ). We previously discovered that the unliganded RARα and γ repress gene transcription via formation of a very specific interface with corepressors [le Maire, Nat Struct Mol Biol, 2010].
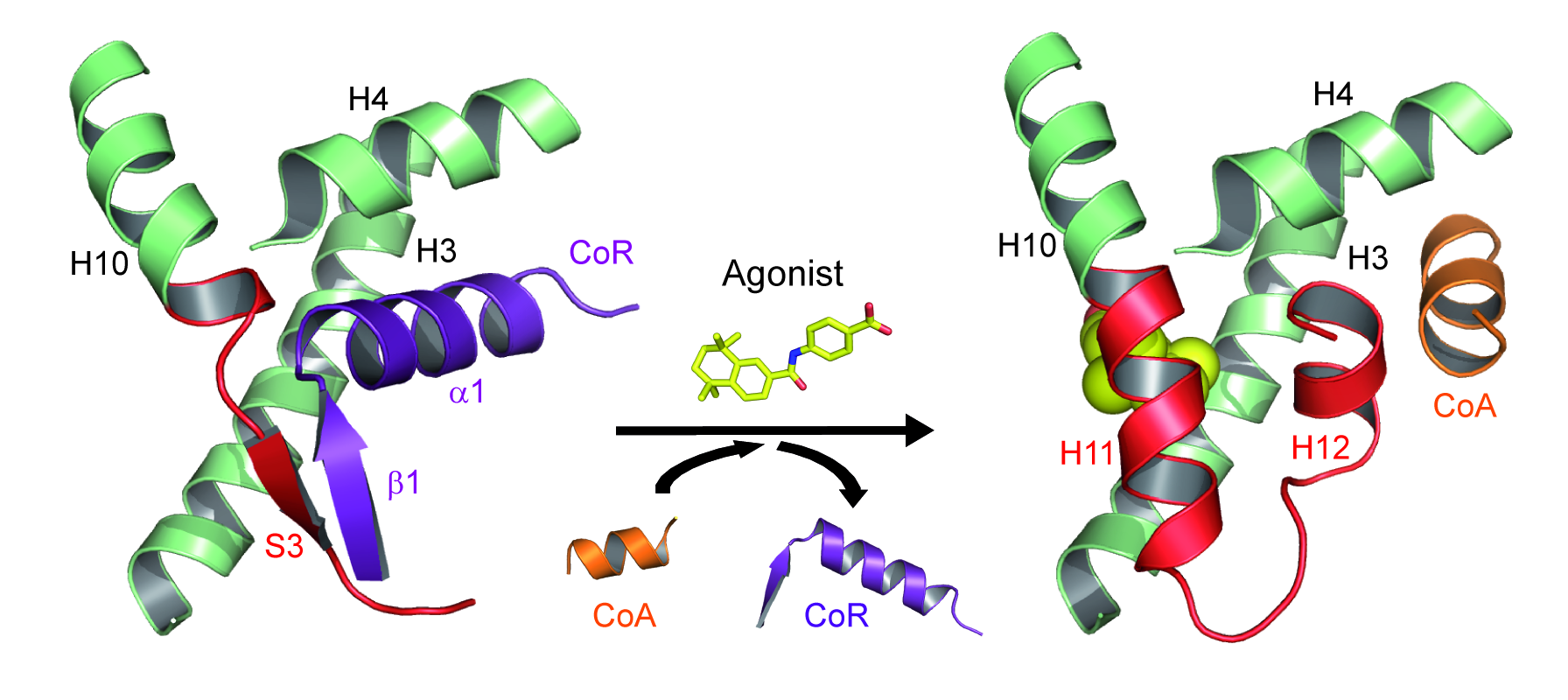
Association and dissociation of coregulators from RAR upon ligand binding (CoA, coactivator ; CoR, corepressor).
More recently, we combined a large set of biophysical and computational methods to show that while the NR interaction domain (NID) of corepressors is mainly disordered, it presents transient but robust intramolecular contacts upon interaction with the heterodimer, indicating that disorder-to-order transitions of both receptors and corepressors are key events in the regulation of RXR–RAR heterodimers [Cordeiro, Structure, 2019] (coll. Team 2). A similar approach is currently being used to investigate the ligand-dependent interaction between the RAR–RXR heterodimer and the NID of the coactivator TIF2 [Senicourt, submitted, bioarchiv]. The fine understanding of these molecular processes at the atomic scale allowed us to insert a mutation into the RARα and γ sequences that specifically disrupts corepressor binding without affecting any other function of the receptors. RARAI396E and RARGI387E were subsequently introduced in mouse to probe the role of RAR-mediated gene silencing in vivo (coll. M Mark/N Ghyselinck). We are currently running genome-wide analyses of genes repressed by RARs through ChIP-Seq and RNA-Seq methods. The interaction sites of the corepressor SMRT in RAR target genes promoters in the absence of ligand or in the presence of various classes of retinoids were identified, as well as the associated epigenetic modifications of chromatin (ongoing work).
We are also interested by the structural and functional characterization of pathological forms of RAR. In particular, acute promyelocytic leukemias (APLs) arise from different chromosomal translocations that create x-RARα fusion proteins, including PML-RARα. Unlike RARα, these x-RARα proteins fail to release properly corepressors in response to ATRA and function as dominant-negative inhibitors of target gene expression and of myeloid cell differentiation. Despite important insights on PML-RARα (and to a lesser extent on other fusion proteins) functioning, the question still remains as to what are the crucial determinants for x-RARα proteins oncogenic activity. Using a set of structural methods, together with cell-based and biophysical assays, we expect to bring new insights into the molecular mechanisms of gene repression mediated by RAR in pathological situations.
Theme 1 also comprises the study of secondary coactivators that are part of coregulatory complexes but not directly bound to receptors. In particular, we studied the human mixed lineage leukemia 5 (MLL5), identified in a multi-subunit complex associated with RARα and thought to play a key role in hematopoiesis, spermatogenesis and cell cycle progression. Having provided compelling evidence that MLL5 is not a functioning enzyme despite its first identification as a member of the histone methyltransferases (HMT) family, we proposed that MLL5 functions as a mediator of gene regulation, not through an HMT activity, but as a structural component ensuring multi-protein complex integrity [Mas-y-Mas, PloS One, 2016].
We are also interested in the evolution of ligand binding activity/specificity in retinoid receptors [Handberg-Thorsager, Sci Adv, 2018 ; Gutierrez-Mazariegos, R Soc Open Sci, 2016 ; Gutierrez-Mazariegos, Endocrinology, 2014] (coll. V Laudet) and in the characterization of selective retinoids that can be used as investigation tools [le Maire, Cells, 2019]. Particularly, many studies have provided conflicting information on the role of RARβ in cancer development so that the availability of RARβ-selective ligands would allow dissecting the specific activities exerted by this receptor. By combining medicinal chemistry, cell biology and X-ray crystallography, we discovered the first full RARβ-selective agonist [Nadendla, PloS One, 2015], whose unexpected binding mode may, in addition, provide a comprehensive set of guidelines for the rational design of RARβ-selective antagonists.
Main collaborators: G Benoit (ENS Lyon), N Ghyselinck/M Mark (IGBMC Illkirch), P Veber (LBBE Lyon), V Laudet (OOB Banyuls/mer), Team 2 (CBS), H de Thé (Collège de France)
Theme 2 : Novel nuclear receptor-based therapeutic strategies
A le Maire, W Bourguet
NRs are important for homeostatic control and for adaptation to environmental variation. They thus participate in molecular mechanisms whose dysregulation is associated with proliferative, reproductive, and metabolic diseases, such as cancer, infertility, obesity, or diabetes. As such NRs are the molecular target of approximately 15% of authorized medications.
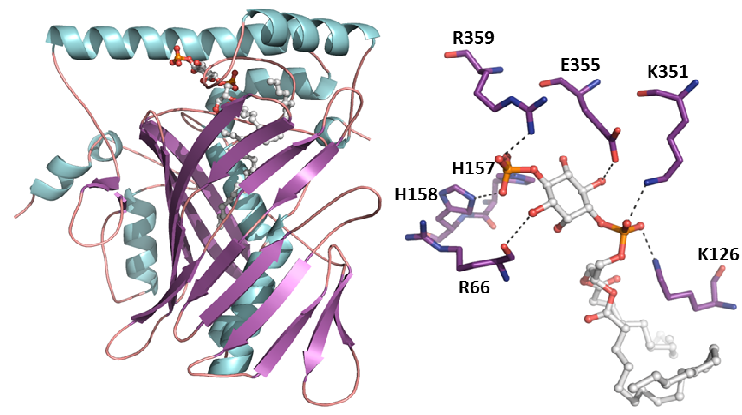
We have examined multiple medically important NR/corepressor aberrant interactions with the objective of designing small molecules or peptidomimetics that could specifically disrupt the undesired interactions. In collaboration with M Privalsky, we described the specific interaction surfaces between pathogenic thyroid hormone receptor β (TRβ) mutants and corepressors, and demonstrated that a ligand (TRIAC) is a potential therapeutic agent for patients suffering from resistance to thyroid hormone (RTHβ) syndromes [Harrus, Thyroid, 2018]. As part of an ANR project (ThyroMUT2 2015-2019) conducted in collaboration with F Flamant and clinicians, we are currently finalizing a similar work on a set of recently described TRα mutants causing a new type of RTHα where we decipher the molecular basis underlying the phenotype variability associated with different THRα mutations [Espiard, J Clin Endocrinol Metab, 2015 ; le Maire, Thyroid, 2020].
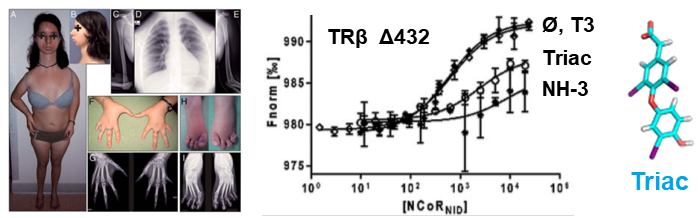
The resistance to thyroid hormone (RTH) syndrome results from the incapacity of corepressors to dissociate from TR upon natural hormone (T3) binding.
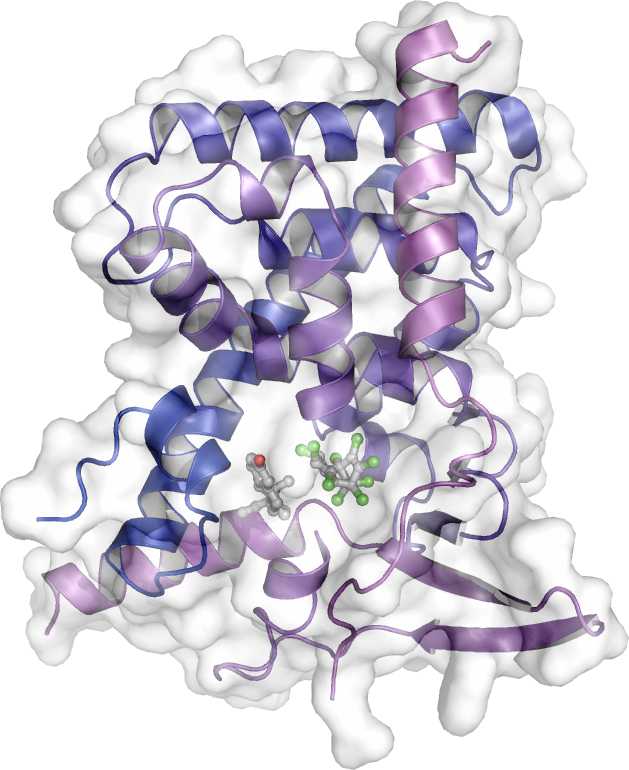
As a main player in drug resistance, the pregnane X receptor (PXR) has also been (and remains) the subject of intense research in our group. Upon activation by many external compounds, including drugs, PXR induces the transcription of genes encoding xenobiotic metabolizing enzymes and efflux pumps that trigger body detoxification. However, this process severely reduces the effectiveness of drugs by shortening their lifetime. In a collaborative work with Team 4, we solved the crystal structure of PXR in complex with an anticancer drug that was recently shown by our collaborator P Balaguer to act as a potent PXR agonist (EC50 < 100 nM). We then rationally designed derivatives that have lost their binding capacity to PXR while retaining high binding affinity for their pharmacological target (work in progress). Another way to prevent premature metabolization of drugs is to inhibit PXR activity. In collaboration with JM Pascussi, M Amblard we rationally designed a bi-functional ligand also referred to as PROTAC (PROteolysis-TArgeting Chimera) that simultaneously binds to PXR and recruits E3 ligases to direct PXR to degradation by the proteasome (ongoing work).
The peroxisome proliferator-activated receptor γ (PPARγ) is a master regulator of adipogenesis and a potent modulator of whole-body lipid metabolism and insulin-sensitivity, making it a valuable pharmaceutical target against obesity and diabetes. Interestingly, it has been recently reported that phosphorylation of PPARγ on S273 correlates with repression of a subset of target genes shown to be dysregulated in obesity. In the aim of identifying novel PPARγ ligands with therapeutic potential, we have described the interaction surface between PPARγ and CDK5 and developed an assay pipeline capable of detecting ligands that physically bind to PPARγ and inhibit the CDK5-mediated phosphorylation without invoking its transcriptional activity and the associated side effects [Ribeiro, Front Endocrinol, 2018 ; Ribeiro, J Struct Biol, 2019; Dias, Front Endocrinol, 2020] (coll. AC Figueira).
Ivermectin (IVM) is the most important broad spectrum antiparasitic drugs used today. Unfortunately, resistance to IVM has appeared in parasites and it seriously jeopardizes the success of current parasite control. By using an original IVM-resistant C. elegans strain, our collaborator A. Lespine showed that the Nuclear Hormone Receptor NHR-8 is a key player in the dynamic development of resistance to IVM. NHR-8 stands at the cross-road of major metabolic pathways in nematodes. We propose to solve NHR-8 structure and to identify its ligands to help deciphering NHR-8 functions and translateing these insights to other target parasitic nematodes with the goal of developing new anti-infectious approaches.
Main collaborators: F Flamant (IGFL Lyon), M Privalsky (UC Davis USA), AC Figueira (CNPEM Brasil), JM Pascussi (IGF Montpellier), M Amblard (IBMM Montpellier), P Balaguer (IRCM Montpellier), A. Lespine (INRAE, Toulouse), Team 4 (CBS)
")
")