Plateau de Résonance Magnétique Nucléaire (RMN)
Responsables scientifiques à contacter : Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser. ou Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser.
Responsable technique : Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser.
Personnel RMN : Hélène Déméné, Philippe Barthe, Nathalie Sibille, Yinshan Yang
Matériels techniques
 |
 |
 |
|
Bruker 800 MHz
4 canaux Sonde 15N-13C TCI
TXI
Avance III 2016
Passeur d'échantillons
|
Bruker 700 MHz
4 canaux 15N-13C TCI + Haute pression
TXI
Avance III 2009
|
Bruker 600 MHz
Sonde BBI + Haute pression
Avance III 2009
|
Résonnance Magnétique Nucléaire RMN :
La RMN est une technique de spectroscopie basée sur les propriétés magnétiques de certains noyaux atomiques (1H, 13C, 15N, 31P, ... ). L'échantillon placé dans un champ magnétique intense va être perturbé par des impulsions radiofréquence. L'enregistrement du retour à l'équilibre des spins permet d'avoir accès à l'environnement chimique des atomes. Ces informations offrent la possibilité d'analyses structurales et dynamiques ainsi que l'étude d'interactions impliquant des macromolécules biologiques.
Principales applications :
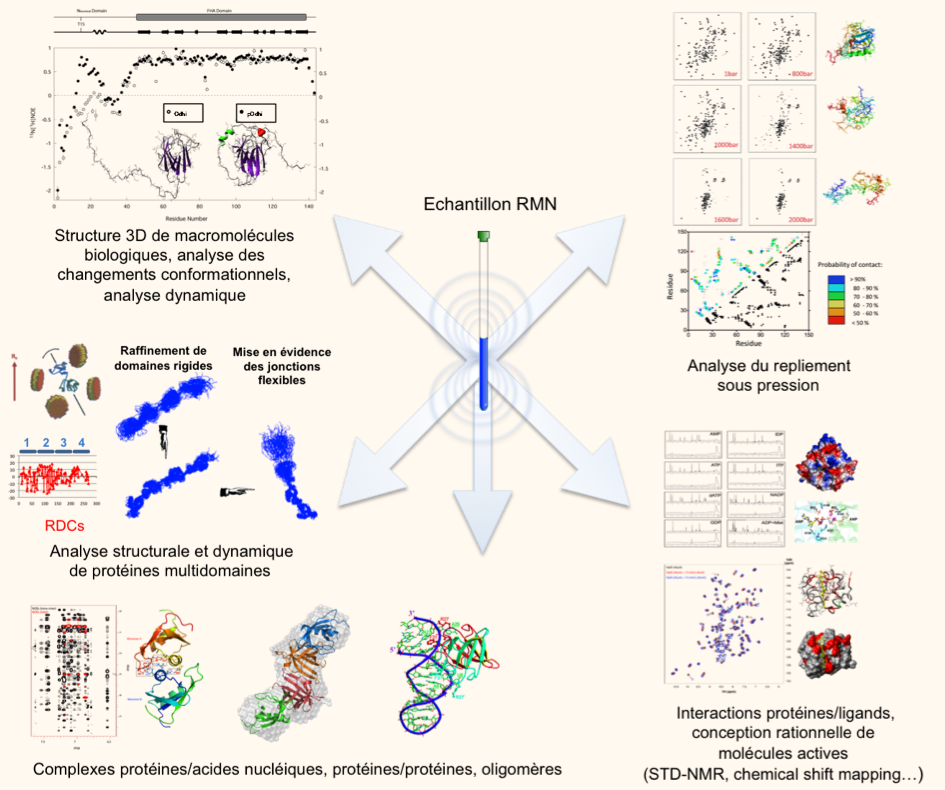
Etude type en RMN
o 1 : Objectif :
Proposer à la communauté scientifique française et européenne des possibilités d'analyse structurales et d'étude d'interaction par RMN pour des protéines et des oligonucléotides.
o 2 : Études d'interactions moléculaires
L'interaction moléculaire entre une macromolécule biologique et un petit ligand peut-être facilement suivie par RMN, en particulier dans le cas de constantes d'affinités faible de l'ordre de 1-1000 micromolaire. Cette étude peut aussi être réalisée sur une bibliothèque de ligands, dans la mesure ou la taille de la bibliothèque est faible (quelques dizaines maximum)
Un marquage isotopique 15N est requis pour cette étude.
L'étude par RMN est faite en haut-champ (700, 600 ou 500 MHz)
Spectres non attribués :
Après une étude de faisabilité, un spectre HSQC est réalisé pour chacun des partenaires potentiels. Le déplacement des signaux de la macromolécule est la signature de l'interaction. Chaque spectre consomme de l'ordre de 100 nanomole de molécule.
Spectres attribués :
L'attribution des spectres permet l'interprétation des résultats en termes structuraux, en particulier quand la structure de la molécule, ou d'un homologue, est déjà connue. Après une étude de faisabilité, un spectre HSQC est réalisé pour chacun des partenaires potentiels. Chaque spectre consomme de l'ordre de 100 nanomole de molécule. Le déplacement des signaux de la macromolécule est la signature de l'interaction, et l'attribution permet de déterminé quelles zones structurales de la macromolécule sont impliquées dans cette interaction.
o 3 : Attribution des spectres de biomolécules
L'attribution du spectres des molécules étudiées (peptides protéines, ADN, ARN) est une étape essentielle pour aller vers l'obtention d'informations structurales. Dans le cas de molécules dont la structure d'homologue est connue, l'attribution permet de vérifier si la molécule étudiée présente le même repliement, et permet d'affiner l'alignement structural.
Les techniques employées s'appuient sur le marquage isotopique en15N et 13C, et dépendent de la taille de la molécules étudiée. Une pré-étude est réalisée qui permet d'évaluer la qualité des spectres et la chance de mener à bien l'étude.
protéine<5 kDa :
Après une étude de faisabilité, le spectre 1H est attribué à partir d'une série d'expériences de RMN-2D : COSY, TOCSY, NOESY, ROESY, 13C-HSQC, 13C-HSQC-TOCSY qui ne nécessitent pas un marquage isotopique. Il est nécessaire pour cette étude de pouvoir atteindre des concentrations de l'ordre du 1mM. Environ 3 micromole de l'échantillon sont nécessaires pour les études de faisabilité et d'attribution.
entre 5 et 10 kDa :
Après une étude de faisabilité, les spectres sont attribués à partir d'une série d'expériences de RMN 2D et 3D double résonance : HSQC, TOCSY-HSQC, NOESY-HSQC qui nécessitent un marquage isotopique en 15N. Il est nécessaire pour cette étude de pouvoir atteindre des concentrations de l'ordre de 0.5mM à 1mM. Environ 1 micromole de l'échantillon sont nécessaires pour les études de faisabilité et d'attribution.
entre 10 et 20 kDa :
Après une étude de faisabilité, les spectres sont attribués à partir d'une série d'expériences de RMN 3D triple résonance : HNCA HNCO HNCACB HNCACO HN(CO)CA CBCA(CO)NH (HACA)CO(CA)NH qui nécessitent un double marquage isotopique en 15N et 13C. Il est nécessaire pour cette étude de pouvoir atteindre des concentrations de l'ordre de 0.5mM à 1mM. Environ 1 micromole de l'échantillon sont nécessaires pour les études de faisabilité et d'attribution.
o 4 : Détermination de structure de biomolécules :
Cette étape nécessite l'attribution préalable des signaux. La structure peut être déterminée par la collecte des données RMN de contraintes.
protéine <5 kDa :
A partir de l'attribution, un jeu de contraintes est extrait des spectres NOESY obtenus et le logiciel CYANA est utilisé pour déterminer la structure en solution de la molécule.
entre 5 et 10 kDa :
Un jeu d'expériences 2D et 3D complémentaires est réalisé : NOESY, TOCSY, TOCSY-HSQC, NOESY-HSQC et à partir de l'attribution, un jeu de contraintes est extrait des spectres obtenus et le logiciel CYANA est utilisé pour déterminer la structure en solution de la molécule.
entre 10 et 20 kDa :
Un jeu d'expériences complémentaires 2D, 3D et 4D est réalisé : NOESY, TOCSY, (h)CCH-tocsy, H(c)CH-tocsy, 15N-TOCSY-HSQC, 15N-NOESY-HSQC, 13C-NOESY-HSQC, HN-NOESY-CH.
A partir de l'attribution, un jeu de contraintes est extrait des spectres obtenus et le logiciel CYANA est utilisé pour déterminer la structure en solution de la molécule.
Microscopies Optiques
Responsable scientifique à contacter : Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser.
Responsables techniques : Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser. , Jean-Bernard Fiche
Matériels techniques
Etude type en Biophysique
o 1 : Objectif
Cette plateforme propose un ensemble d'approches basées sur la spectroscopie de fluorescence applicables à des études d'interactions entre biomolécules et de dynamique structurale (repliement, assemblage, diffusion, fluctuations, changements conformationnels). Contactez Emmanuel Margeat ou Caroline Clerte pour discuter des possibilités d'utilisation de la plateforme et de la conception des expériences.
Ci dessous une présentation des différentes techniques utilisées au CBS (bientôt disponible).
")
")