")
")
Regulation of gene transcription by retinoic acid receptor heterodimer and its coregulators
Regulation of gene transcription by retinoic acid receptor heterodimer and its coregulators (PIs: N. Sibille & P. Bernadó)
People Involved: N. Sibille, P. Bernadó, F. Allemand, A. Fournet, T. Cordeiro, L. Sénicourt, A. Sagar
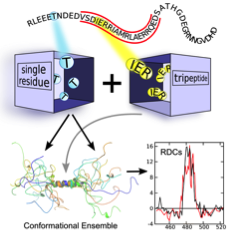
Nuclear Receptors (NRs) are transcription factors with a direct role in regulating the expression of hormone-response genes. Agonist binding to NRs triggers transcription through a cascade of events that is initiated by the exchange of the repression complex by the activation one. The assembly of the two large complexes is mediated by intrinsically disordered coregulator proteins that, simultaneously, also interact with the NRs. Coregulators are named coactivators or corepressors when they mediate the activation or repression complexes, respectively. In collaboration with A. le Maire, W. Bourguet and P. Germain we have dissected the structural bases of the interaction between both families of coregulators with the Retinoic Acid Receptor heterodimer (RAR/RXR). Funded by the Labex EpiGenMed, we have studied the interaction of the intrinsically disordered NCoR (corepressor) and TIF2 (coactivator) with RAR/RXR, which form highly dynamic and malleable complexes.
![]()
We reached to the following conclusions for the corepressor NCoR (Cordeiro et al., 2019): (i) Small ligands induce disorder to order transitions in the C-
terminal helix of the Ligand Binding Domain (LBD) of both RAR and RXR, and they occur
independently of the presence of coregulator proteins.
(ii) We demonstrated that the NR binding region of NCoR is intrinsically disordered, but it encompasses three highly conserved regions that are partially structured that are linked to RAR/RXR recognition. (iii) The NCoR complex with RAR/RXR is highly plastic as N-CoR is bound to the heterodimer in a singly- and a doubly-bound form (Fig. 3). The relative populations of these complexes can be modulated with small ligands and point mutations.
The main results derived from the study of the coactivator TIF2 are (Sénicourt et al.,
in preparation): (i) We demonstrated by NMR that the NR binding region of Tif2 is intrinsically disordered, but it encompasses three highly conserved regions that are partially structured. (ii) In addition of the three well-known NR interacting sites, we have highlighted importance of a flanking region in strengthening the interaction with RAR/RXR. Initially identified by NMR, this flanking region has been successfully co-crystalized with the nuclear receptor, displaying a helix-turn-helix motif. Further studies will be performed to dissect the relevance of this interaction.
These results provide the structural and functional clues for the regulation of gene transcription and suggest mechanistic pathways for the transition from basal (inactive) state to the transcriptionally active one.
Main collaborators: A. LeMaire, P. Germain, W. Bourguet, A. Barducci (CBS, Montpellier)
Grant: Labex EpiGenMed