RMN haute pression
C. Roumestand, N. Declerck, P. Barthe, C. Dubois
The team has done pioneering works in the field of High-Pressure (HP) NMR. Through a tight collaboration with Catherine A. Royer, an expert in HP Biophysics and former director of the CBS, the group of C. Roumestand was the first French NMR group to develop High-Pressure NMR methodology applied to protein folding analysis (for reviews see Roche et al., Prog. Nucl. Magn. Reson. Spectrosc. 2017; Roche et al., Meth. Enzymol. 2019). Protein folding constitutes still a very active research field for the team, with several on-going projects. Besides, the group is involved in ambitious projects concerning Microbial adaptation to high Pressure.
High-Pressure NMR and Protein Unfolding
C. Roumestand, P. Barthe, C. Dubois
High hydrostatic pressure (HHP) is a very useful reversible perturbation method for exploring the thermodynamics of the folding/unfolding equilibrium of biomolecules. When combined to NMR spectroscopy, HHP offers the possibility to monitor at a single residue level the structural transitions occurring upon protein denaturation.
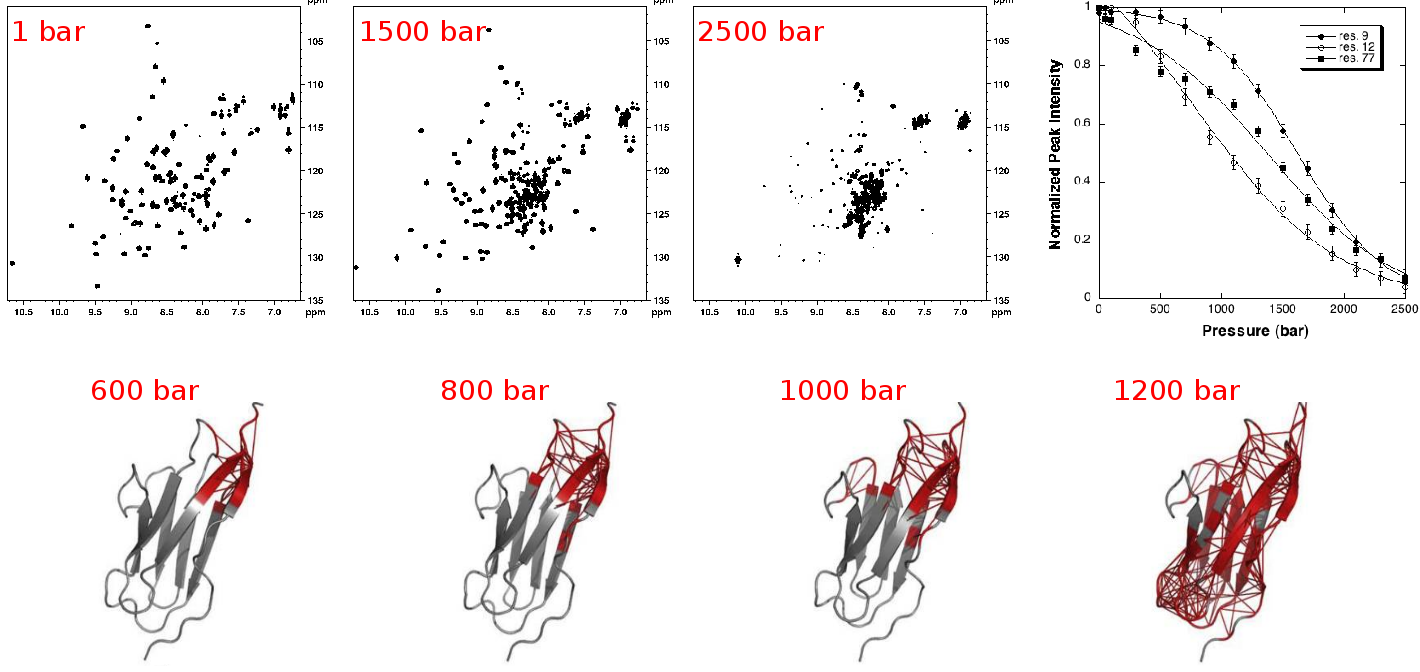 |
| NMR detected high pressure unfolding of Titin I27 single-module. Upper panels: examples of [1H-15N] HSQC NMR spectra at different pressures and residue-specific denaturation curves obtained for 3 residues exhibiting distinct unfolding profiles. Lower panels: ribbon representations of Titin I27 solution structure showing in thick red the contacts that are weakened at the indicated pressures. Adapted from Herrada et al., Biophys J, 2018. |
We use high-pressure NMR to reach a detailed structural and energetic characterization of the folding process of proteins such as the leucine-reach repeat protein pp32 or immunoglobulin-like domains from the dengue virus envelope protein or the protein titin (single- or bi-domains) from sarcomeres. Special emphasis has been put on the study of folding cooperativity inside a single domain (pp32 leucine-reach repeat domain) or between two domains in modular protein (titin). Similar studies are on-going, trying to establish the relationships between protein sequence, protein topology, protein 3D structure and folding routes.
We also use HP-NMR to address fundamental issues regarding protein conformational landscapes or the links between specific thermodynamic parameters and biological properties, such as between thermal expansion and conserved pattern of interactions in proteins, or between protein-ligand binding volumes and affinity.
Collaborations: F. Rico (Marseille); C. Royer (RPI, Troy, USA); J. Roche (Iowa State Univ., USA); Y. Kuroda (Tokyo University, Japan)
References: Dellarole et al., J Am Chem Soc, 2015; Fossat et al., Biophys J, 2016; Toleikis et al., J Phys Chem B, 2016; Herrada et al., Biophys J, 2018; Roche et al., Prog Nucl Magn Reson Spectrosc., 2017; Meth Enzymol, 2019; Saotome et al., Biomolecules, 2019
Microbial Adaptation to High-Pressure
Adaptation to life under HHP
C. Roumestand
|

Hydrothermal vents in the Mariana trench: the cradle of life ?
|
|
Adaptation to life under high hydrostatic pressure is a trait shared between the first microbial cells and today's piezophiles (HP-adapted). Understanding the basis of this adaptation in contemporary cells is essential to understand the origin of life. To identify these adaptive signatures, we propose to study the molecular evolution of key proteins in piezophilic and piezosensitive Archae, in collaboration with Microbiologists specialized in extremophiles (P. Oger, ENSA Lyon) and a Physicist specialized in HP-SANS measurements (J. Peter, ILL Grenoble) |
Pathogens Adaptation to High Pressure
N. Declerck, C. Roumestand
| |
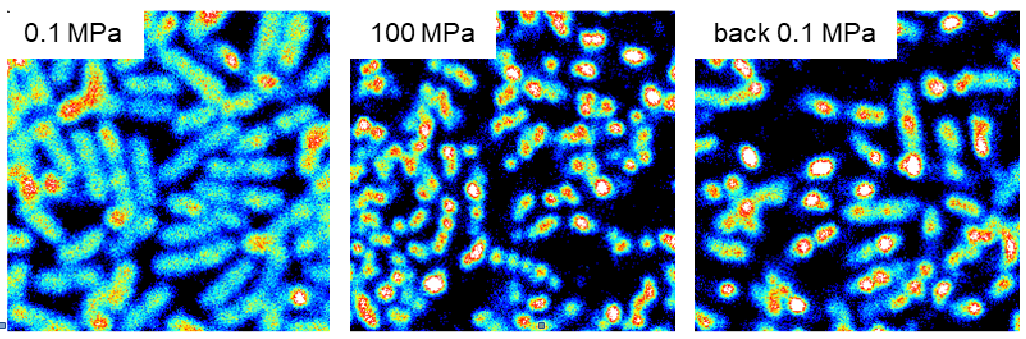
Fluorescence intensity images (13x13 µm) of E. coli cells expressing GFP-Mrr at atmospheric pressure (0.1 MPa), under high pressure (100 MPa) and after pressure is released (back 0.1 MPa).
|
|
Resistance to high pressure can be acquired by potential human pathogens exposed to barotromatic treatments such as those now routinely used in the food industry as non-thermal sterilization processes. In E. coli, the Mrr endonuclease is responsible for the activation of the SOS reponse after a HP shock and mrr mutations conferring resistance to pressure up to 2 GPa can be selected in only a few generation. We are currently investigating the oligomeric switch on which rely Mrr HP-induced activation, in collaboration with A. Aertsen at KU Leuven (Belgium) and C. Royer at RPI (Troy, USA) who developed a microscopy setup for fluorescence correlation measurements under high pressure.
Pathogens & Infectious Diseases
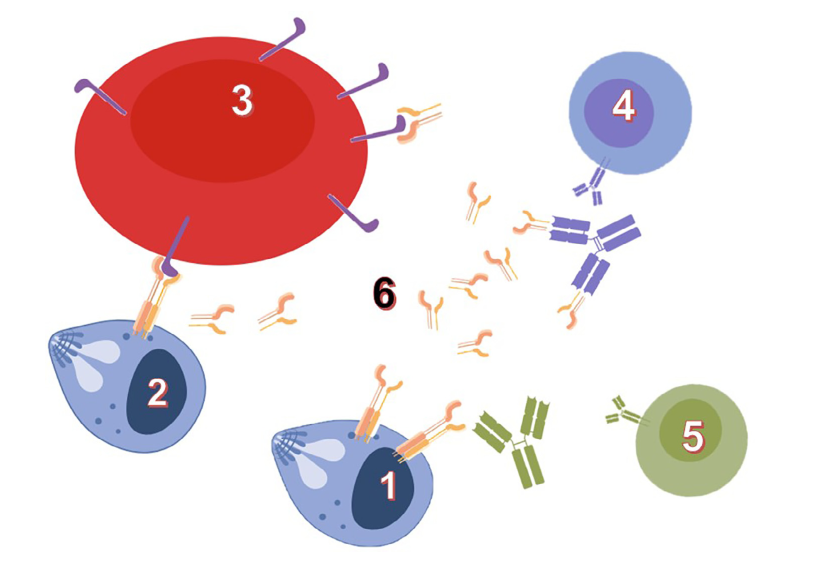
Mechanistic and structural bases of recognition between avirulence proteins (AVR) and resistance proteins (R).
André Padilla & Karine de Guillen

The molecular bases of plant resistance against pathogenic fungi aggressions are poorly understood. This immunity is based on two levels defence system. The first line involves the detection of microbial molecules, such as flagellin or components of the cell wall of the pathogenic microorganism, leading to resistance. To avoid this first level the fungus secretes, during infection, small proteins called avirulence (AVR), which are considered as key elements of fungal pathogenicity but whose functions are largely unknown. The recognition of these plants pathogenic effectors is triggered by resistance proteins (R), which constitute the second layer of defence. This recognition leads to a "hypersensitive" response, which is characterized by localized cell death, avoiding plant colonization by the pathogen.
The fungus Magnaporthe oryzae is the major pathogen of rice. The disease triggered by this fungus is called rice blast. It is responsible for large economic losses and is a major threat to food security. In recent years collaboration with the team of T. Kroj (INRA-Montpellier BGPI) was established. Our collaborators have demonstrated the interaction between avirulence protein of M.oryzae AVR-Pia and AVR-CO39 and resistance proteins RGA4-RGA5 (Cesari et al, Plant Cell, 2013, Ribot et al, The Plant Journal 2013). The production of recombinant avirulences proteins allowed us to determine their three-dimensional structures by NMR. These recent findings raise several questions:
- How are AVR proteins specifically recognized by "their" R protein?
- What are the key amino acids involved in the interaction?
- How is AVR recognition linked to defence activation in R proteins hetero pair?
Validation of structures (AVR and R-domains) by functional studies will help us to understand the mechanisms of recognition and co-evolutions between AVR and R
Interaction between fungus Avr proteins and plant R proteins. (adapted from Liu et al 2014)
External collaboration: T. Kroj INRA-BGPI – Montpellier – France
Structure and dynamics of RYMV viral encoded P1 protein
Hélène Déméné & Yinshan Yang
The P1 protein is a crucial protein of the RYMV (Rice Mottle Yellow) virus that infects the most productive rice plants in Africa. Using an integrative approach combining X-Ray and NMR spectroscopy at the CBS, we have characterized the structure and revealed the dynamics of the RYMV P1 protein that are linked to the mode of viral activation.
External collaboration: Florence Vignols and Christophe Brugidou (IRD)
Structure of Major Antigens from Apicomplexa.
Christian Roumestand & Yinshan Yang.
Babesiosis (formerly known as piroplasmosis) is a tick-borne disease caused by the intraerythrocytic development of protozoa parasites from the genus Babesia. Like Plasmodium falciparum, the agent of Malaria, or Toxoplasma gondii, responsible for human toxoplasmosis, Babesia belongs to the Apicomplexa family. Babesia canis is the agent of the canine babesiosis, while Babesia divergens infects essentially bovine. The identification and characterization of parasite surface proteins represent major goal, both to understand the molecular bases of Apicomplexa invasion process and for the potential as vaccines of such antigens. Indeed, it has been shown that the GPI membrane anchored protein Bd37, a 37 kDa antigenic adhesion protein from B. divergens, was able to induce complete protection against various parasite strains. Its orthologue in B. canis, Bc28.1, has been described as a 28 kDa membrane protein GPI anchored at the surface of the merozoite. We have defined the erythrocyte binding function of these two proteins and determined their high-resolution solution structure using NMR spectroscopy (Delbecq et al., 2008: Yang et al., 2012). Surprisingly, although these proteins are thought to play a similar role in the adhesion process, the structure of Bc28.1 (see Figure) appears unrelated to the structure of Bd37. Site-directed mutagenesis experiments also suggest that the mechanism of the interaction with erythrocyte membrane could be different for the two proteins. The resolution of the structure of these adhesion proteins represents a milestone for the characterization of the parasite erythrocyte binding and its interaction with the host immune system.

Solution structure of adhesion proteins from Babesia.
(Left) Bd37 from B. divergens; (Right) Bc28 from B. canis
External Collaborations:
· R. Cerdan – DIMNP – Montpellier – France
· S. Delbecq – University Montpellier I – Montpellier – France
")
")