")
")
RMN haute pression
C. Roumestand, N. Declerck, P. Barthe, C. Dubois
The team has done pioneering works in the field of High-Pressure (HP) NMR. Through a tight collaboration with Catherine A. Royer, an expert in HP Biophysics and former director of the CBS, the group of C. Roumestand was the first French NMR group to develop High-Pressure NMR methodology applied to protein folding analysis (for reviews see Roche et al., Prog. Nucl. Magn. Reson. Spectrosc. 2017; Roche et al., Meth. Enzymol. 2019). Protein folding constitutes still a very active research field for the team, with several on-going projects. Besides, the group is involved in ambitious projects concerning Microbial adaptation to high Pressure.
High-Pressure NMR and Protein Unfolding
C. Roumestand, P. Barthe, C. Dubois
High hydrostatic pressure (HHP) is a very useful reversible perturbation method for exploring the thermodynamics of the folding/unfolding equilibrium of biomolecules. When combined to NMR spectroscopy, HHP offers the possibility to monitor at a single residue level the structural transitions occurring upon protein denaturation.
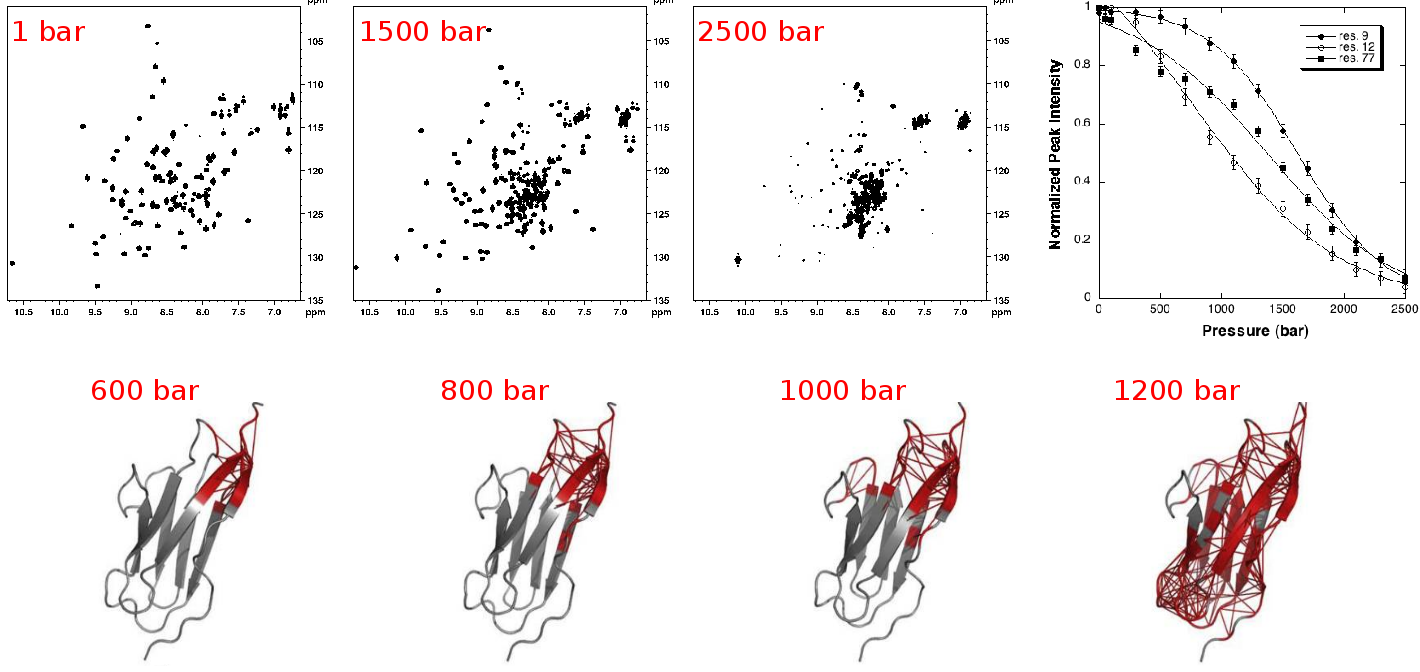 |
| NMR detected high pressure unfolding of Titin I27 single-module. Upper panels: examples of [1H-15N] HSQC NMR spectra at different pressures and residue-specific denaturation curves obtained for 3 residues exhibiting distinct unfolding profiles. Lower panels: ribbon representations of Titin I27 solution structure showing in thick red the contacts that are weakened at the indicated pressures. Adapted from Herrada et al., Biophys J, 2018. |
We use high-pressure NMR to reach a detailed structural and energetic characterization of the folding process of proteins such as the leucine-reach repeat protein pp32 or immunoglobulin-like domains from the dengue virus envelope protein or the protein titin (single- or bi-domains) from sarcomeres. Special emphasis has been put on the study of folding cooperativity inside a single domain (pp32 leucine-reach repeat domain) or between two domains in modular protein (titin). Similar studies are on-going, trying to establish the relationships between protein sequence, protein topology, protein 3D structure and folding routes.
We also use HP-NMR to address fundamental issues regarding protein conformational landscapes or the links between specific thermodynamic parameters and biological properties, such as between thermal expansion and conserved pattern of interactions in proteins, or between protein-ligand binding volumes and affinity.
Collaborations: F. Rico (Marseille); C. Royer (RPI, Troy, USA); J. Roche (Iowa State Univ., USA); Y. Kuroda (Tokyo University, Japan)
References: Dellarole et al., J Am Chem Soc, 2015; Fossat et al., Biophys J, 2016; Toleikis et al., J Phys Chem B, 2016; Herrada et al., Biophys J, 2018; Roche et al., Prog Nucl Magn Reson Spectrosc., 2017; Meth Enzymol, 2019; Saotome et al., Biomolecules, 2019
Microbial Adaptation to High-Pressure
Adaptation to life under HHP
|
Hydrothermal vents in the Mariana trench: the cradle of life ? |
Adaptation to life under high hydrostatic pressure is a trait shared between the first microbial cells and today's piezophiles (HP-adapted). Understanding the basis of this adaptation in contemporary cells is essential to understand the origin of life. To identify these adaptive signatures, we propose to study the molecular evolution of key proteins in piezophilic and piezosensitive Archae, in collaboration with Microbiologists specialized in extremophiles (P. Oger, ENSA Lyon) and a Physicist specialized in HP-SANS measurements (J. Peter, ILL Grenoble) |

Pathogens Adaptation to High Pressure
|
|
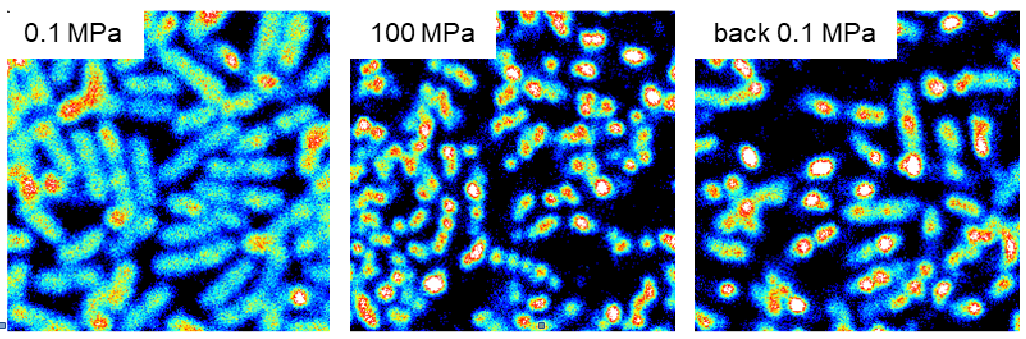
Resistance to high pressure can be acquired by potential human pathogens exposed to barotromatic treatments such as those now routinely used in the food industry as non-thermal sterilization processes. In E. coli, the Mrr endonuclease is responsible for the activation of the SOS reponse after a HP shock and mrr mutations conferring resistance to pressure up to 2 GPa can be selected in only a few generation. We are currently investigating the oligomeric switch on which rely Mrr HP-induced activation, in collaboration with A. Aertsen at KU Leuven (Belgium) and C. Royer at RPI (Troy, USA) who developed a microscopy setup for fluorescence correlation measurements under high pressure.