")
")
rVAP: Recombinant Vaccines Against Parasites
Vaccines against parasitic diseases remain a challenge in terms of target identification, antigen production and formulation to obtain efficient vaccine. Vaccines based on recombinant antigenic proteins constitute an interesting alternative strategy to inactivated or live vaccines but their development is often more difficult.. Aside such recombinant vaccines, companion products are needed to make diagnosis and assess the risk of disease, manage the vaccination scheme in human or animal populations, and evaluate the protection status of individuals. Such management tools are particularly needed for veterinary products and sanitary procedures. . For instance, a vaccine that allows the Differentiating Infected from Vaccinated Animals (DIVA vaccine) could be mandatory for animal exportation. Moreover, the need of Correlate of Protection (CoP) assays for human populations is increasing, as the immunological status of individuals is one of the most important parameter in public health management in case of epidemic diseases.
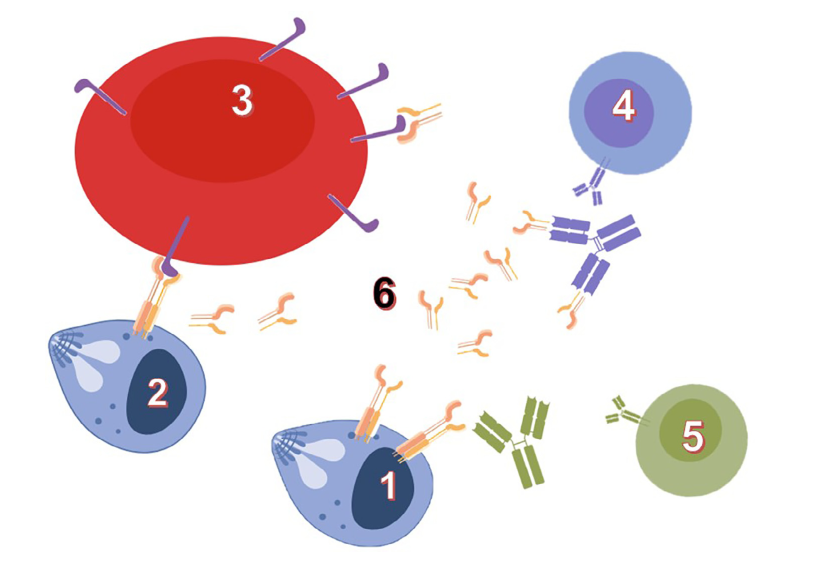
The identification and development of vaccines and diagnostic tools take advantage of deep basic research on host-parasite interfaces. Our model system, Babesia divergens, an intraerythrocytic Apicomplexa parasite transmitted to bovines by ticks, illustrates such potential: an established vaccine system based on a recombinant protein (BD37), a vaccine antigen switching between protective or non-protective induced responses, and investigation levels ranging from molecular structures to experiments in live animals. To address questions about mechanisms of host cell invasion and host immune system escape, Babesia genus offers original and powerful models. Our previous studies highlighted the role of glycosylphosphatidylinositol (GPI)-anchored proteins in "invasion and escape" mechanisms. GPI-anchored proteins are produced as membrane-bound antigens by the parasites, and shed as soluble proteins in the extracellular space (e.g. host blood circulation) Our hypothesis is that the antigen physical state (soluble or membrane-bound) is modulating molecular interactions with the host, and that these interactions drive the immune system toward a protective or non-protective response.
Our current research aims at establishing this protein-embedded immune system escape mechanisms by omparative analysis of GPI-anchored proteins in various Babesia-host models (human, bovine, canine and rodent). This could foster the identification of general principles and methods to decipher parasite invasion and escape mechanisms as well as the influence of GPI-anchors on the recognition of antigenic proteins. Together with an increased knowledge of host-parasite interactions at molecular level, outcomes of these researches provide solutions for animal and human babesiosis studies and management (vaccines, diagnostic tools...). Rational vaccine design against other pathogens (Apicomplexa or not) could also benefit from such knowledge.
 |
|
Glycosylphosphatidyl-anchored proteins (GPI-AP) mediate host-parasite interactions according to their physical state: Wieser et al., Int J Parasitol. 2019; 49(2):175-181: DOI: 10.1016/j.ijpara.2018.12.002 |