")
")
G. Labesse, J-L. Pons, J. Gracy, V. Moreau, R. Salgado Ferreira, M. Schneider
Structural bioinformatics aims at analyzing and predicting protein structure from sequences. We develop tools in order to improve the speed and the quality of the theoretical models one can build. In addition we are implementing an integrated interface connecting protein structure modeling and ligand screening.
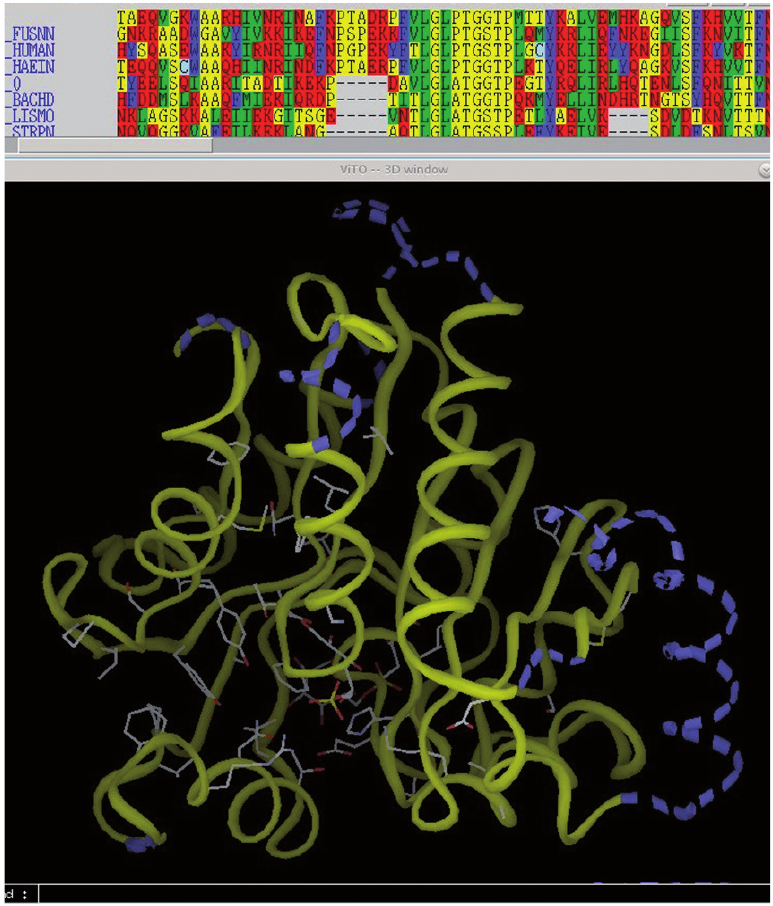
3D visualization and sequence editing using VITO for modeling distantly related proteins
Fold-recognition has been shown to be highly efficient to detect even weak similarities (15-25% of sequence identity). Nevertheless proper alignment and correct model building from distantly related tem-plates still require significant expertise and work. Implementing new refinement approaches is necessary. In parallel, comparative docking has shown an unexpected robustness even at low level of sequence conservation and allowed functional annotation. We now focus our methodology development to improve ligand docking into multiple conformations.
Collaborations : P. Tuffery (Paris Diderot, Paris)
References : Pons & Labesse, NAR, 2009 ; Martin et al., NAR, 2006