Responsables scientifiques à contacter : This email address is being protected from spambots. You need JavaScript enabled to view it.
Responsable technique : This email address is being protected from spambots. You need JavaScript enabled to view it.
Etude type en cristallographie
o 1 : Objectif
Proposer à la communauté scientifique internationale des possibilités d'analyse structurales et d'étude d'interaction par cristallographie pour des protéines et des oligonucléotides.
o 2 : Cristallogenese
Nous avons la possibilité de tester plus de 3600 conditions initiales différentes de cristallisation réparties dans 38 kits (Deep Well au format 96), complétés par 4 kits de détergents (4 x 96) et 3 kits d'additifs (3 x 96) pour affinement. Les techniques de cristallisation sont essentiellement de la diffusion de vapeur (gouttes assises ou suspendues) mais également liquide-liquide ou sous huile.

Chaque piste est affinée (conditions initiales, additifs) avec le robot Hamilton (format 96) ou manuellement (format 15 ou 24)

Les tests de cristallisations sont réalisés avec des gouttes de 0.1 µl. La consommation est de 12ul par boites (kit), il faut donc environ 240 ul pour une étude complète (20 kits). Les 18 autres kits étant pour l'affinement (mono précipitant) ou sprécialisés (Acide nucléique, Membranaire). La protéine doit être pure (sur gel SDS-page) et à une concentration (en moyenne) de 10 mg/ml. Cette concentration initiale est susceptible d'être ajustée (entre 5 et 20 mg/ml) en fonction de la solubilité de la protéine (rapport gouttes précipitées sur gouttes claires) après le premier kit.
Au terme :
- Nous avons un mono cristal d'une taille suffisante pour être manipulé (10 µm). Un test de diffraction est alors réalisé au laboratoire ou à l'ESRF (synchrotron, Grenoble). En fonction de la qualité difractive, la suite sera soit l'affinement (kits suivants, additifs, seeding..), soit la collecte et la résolution de structure.
- Pas de cristaux analysables mais quelques pistes (cristaux maclés ou trop petits, précipités cristallins, oursins, aiguilles...). Dans ce cas, nous tentons une optimisation à partir de chaque condition initiale (additifs, seeding...).
- Aucun cristaux analysable ni de piste. Une réunion avec les parties concernées est alors à envisager pour évoquer le besoin d'analyse de la protéine (polydispersité, protéolyse ménagée si multi domaines ou destructuration partielle...) ou opter pour une résolution par RMN si le poids de la protéine est inférieur à 40 kD.
Le protocole général d'une étude est schématisé sur la figure suivante :
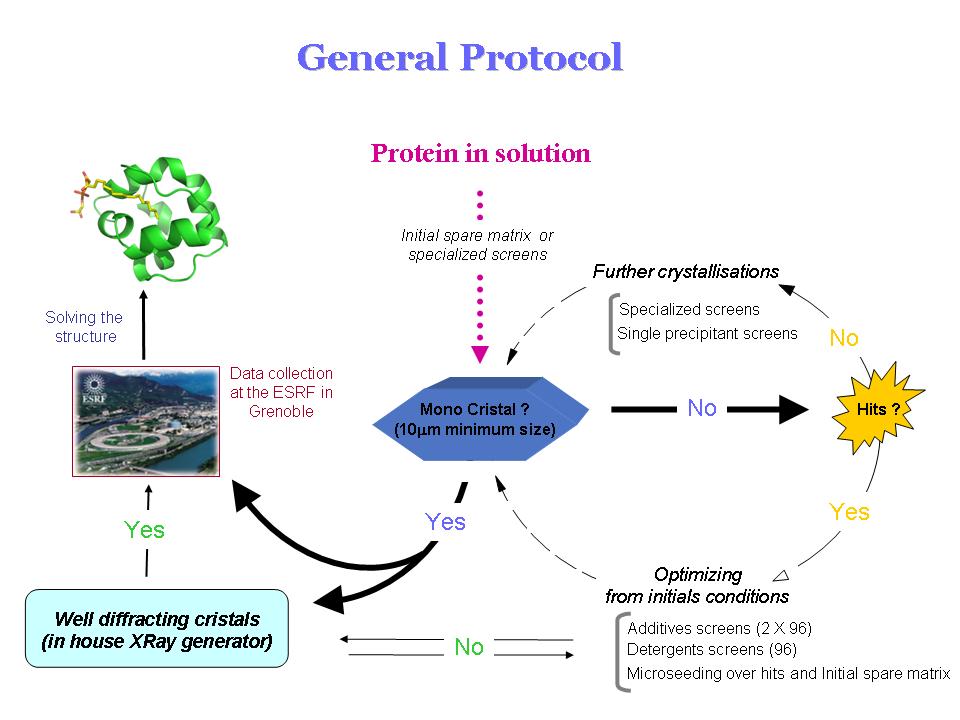
o 3 : Détermination de la structure
La détermination de la structure ne sera engagée qu'après l'obtention d'un jeu complet de données à une résolution d'au moins 3,5 A. Succinctement, la structure sera résolue soit par :
- Remplacement moléculaire si une structure d'une protéine homologue existe (recherchée par bioinformatique).
- Remplacement isomorphe ou diffusion anomale (selon l'élément) en utilisant des dérivés d'atome lourd obtenus soit in vivo (séléno-méthionine, séléno- cystéine) soit par trempage des cristaux avec une solution d'atome lourd (Hg,Pt..)
- Méthode directe si la résolution est > à 1.2 A (extrêmement rare).