Get out of here, let me get on it ! The G protein and the beta-arrestiin
Arginine-vasopressin (AVP) is known as the antidiuretic hormone, and it regulates a vital function of our body: water homeostasis. It plays a role in the kidney at the basolateral membranes of the principal cells of the distal collecting duct of the nephron where it directly interacts with the V2 receptor subtype (V2R), which is a typical G protein-coupled receptor (RCPGs). Upon interaction, AVP activates a signaling pathway leading to fusion of aquaporine water channels to the apical membrane of the cells and consequently to water reabsorption from urine to blood. Coupling of the V2R to the Gs protein constitutes a first key step of the cellular response. But that’s not all! The receptor is not "faithful" to the G protein because it also interacts with ß-arrestins which cause its internalization/desensitization and then activate other signaling pathways involved in particular during cell growth and differentiation. The two canonical signaling partners are two facets of effective regulation of antidiuretic function.
So how does the V2 receptor “choose” between G protein and ß-arrestin? A real Cornelian dilemma… There is a particular interest in answering this question since the development of V2R ligands selective for one pathway or another (biased ligands) presents an important interest to improve the efficiency of future therapeutic molecules, while decreasing their side effects.
After solving the structure of the AVP-bound V2 receptor-Gs signaling complex, the team of Bernard Mouillac and Sébastien Granier at the Institute of Functional Genomics (IGF), in collaboration with the team of Patrick Bron at the Center for Structural Biology (CBS) has now determined the three-dimensional (3D) structure of the AVP bound V2R- ß-arrestin1 complex using cryo-electron microscopy (publication link). It is therefore a work 100% done in Montpellier but also a French first. Today, only 5 structures of signaling complexes of a GPCR coupled to an arrestin have been described: those involving rhodopsin, the beta1-adrenergic receptor, the M2 muscarinic acetylcholine receptor, the NTSR1 receptor of neurotensin and therefore the AVP V2 receptor.
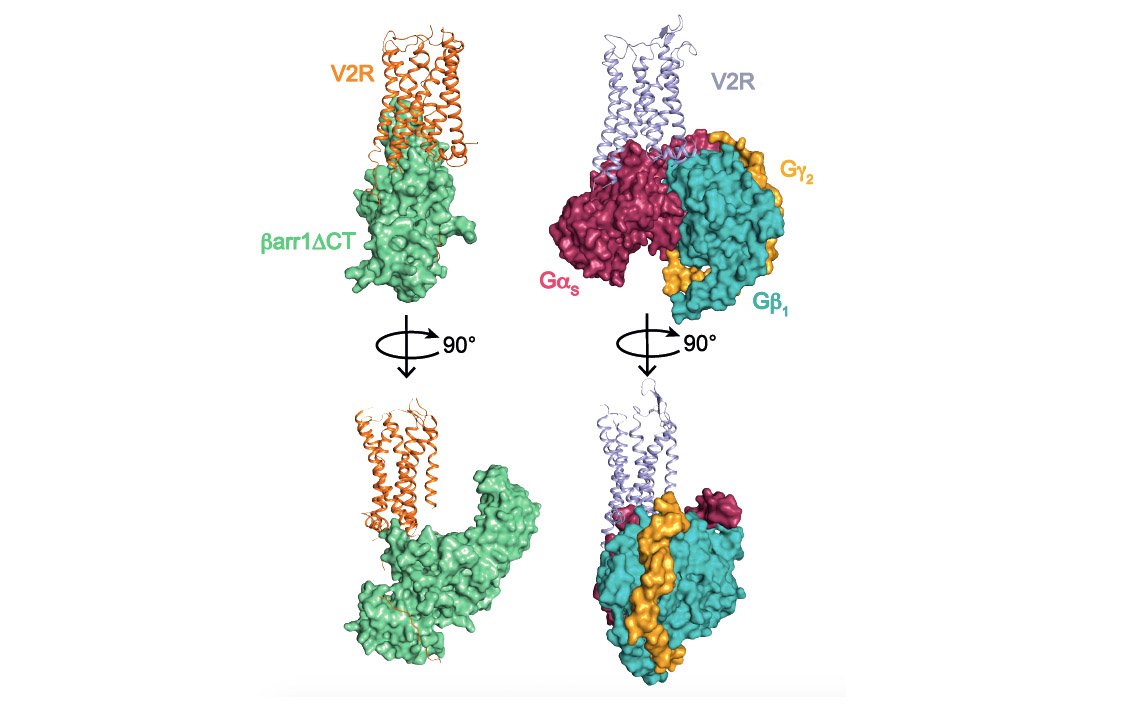
This structural biology approach combined with molecular pharmacology and modeling/molecular dynamics approaches, made it possible to determine the general architecture of the complex and in particular to define the interface between the receptor and ß-arrestin (see associated figure). Several surprises were revealed. First of all, the orientation of ß-arrestin1 with respect to V2R is totally atypical compared to that of arrestins linked to other GPCRs. This positioning highlights the very dynamic character of these signaling complexes and their great structural variability despite a conserved general conformation. Then, the V2R-ß-arrestin1 interface is original because it involves all the intracellular domains of the receptor. It is therefore a specific combination of the conformation of V2R and ß-arrestin1 that explains the originality of the interaction. Finally, the Gs protein and ß-arrestin1 interact with the same intracellular "cavity" of the V2R and are therefore competitive with each other for this binding site. The comparison of the two structures (see associated figure) makes it possible to understand how ß-arrestin "stops" the signal associated with the G protein by taking its place (get out of here, let me get on it), a phenomenon responsible for internalization and desensitization of the receptor.
V2R is a major therapeutic target for treating water metabolism disorders (hyponatremia consecutive to heart failure, hypertension, liver cirrhosis) and voiding disorders (incontinence). In addition, numerous mutations of the receptor are responsible for two rare genetic diseases with opposite clinical outcomes: 1/ congenital nephrogenic diabetes insipidus (cNDI) due to "loss of function" mutations associated with an inability of the patients to concentrate their urine, 2/ nephrogenic syndrome of inappropriate antidiuresis (NSIAD) linked to constitutively active mutations associated to excessive water loading and hyponatremia. V2R is also an essential target for treating certain forms of polycystic kidney disease, a much more frequent disease generally leading to renal failure. In the future, the complete knowledge of the atomic details of the different conformations of the V2 receptor, both active (in the presence of agonists and signaling partners) and inactive (in the presence of antagonists) will guide the rational development of new molecules leading to improved therapies. This research perspective is crucial when dealing with diseases that are difficult to manage and overwhelming for patients.
Publication link:
Structure of the vasopressin hormone-V2 receptor-b-arrestin1 ternary complex.
J. Bous#, A. Fouillen#, H. Orcel, S. Trapani, X. Cong, S. Fontanel, J. Saint-Paul, J. Lai-Kee-Him, S. Urbach, N. Sibille, R. Sounier, S. Granier*, B. Mouillac*, P. Bron*.
# These authors contributed equally to this work *corresponding authors
Science Advances, 2022
Comparison of V2 receptor signaling complexes involving ß-arrestin1 (left) and Gs protein (right). The three subunits a, b and g of the G protein are represented. A truncated version (barr1DCT) of b-arrestin1 was used in this study.
The structure of pathogenic huntingtin exon-1 defines the bases of its aggregation propensity
Link to publication: https://www.nature.com/articles/s41594-023-00920-0
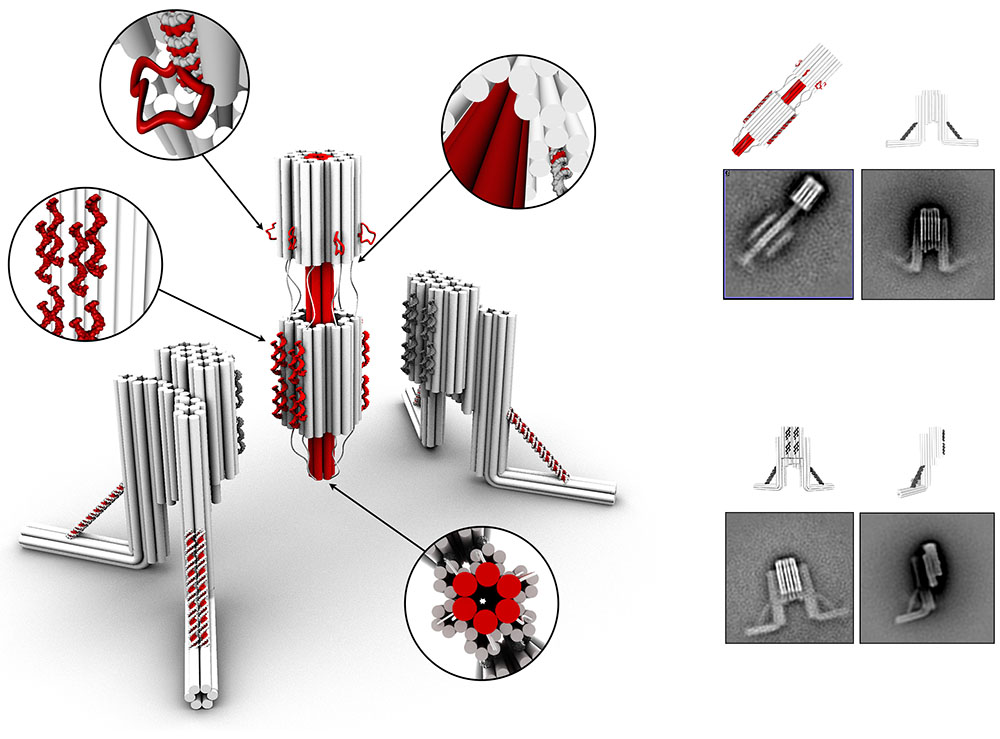
Huntington’s Disease (HD) is a deadly neurodegenerative disorder caused by an expansion of the CAG codons in the first exon of the HTT gene that, after translation, results in an extended poly-glutamine (poly-Q) tract in the N-terminal region of the protein huntingtin (httex1). Only individuals with more than 35 consecutive glutamines in this track develop HD. The structural changes occurring to httex1 when increasing its length beyond this pathological threshold remain poorly understood, precluding a molecular understanding of the pathology. The high-resolution structural investigation of httex1 had been considered as impossible due to its intrinsic flexibility, which preclude the use of X-ray crystallography and cryo-Electron microscopy, and the strong compositional bias that hampers the use of traditional NMR approaches. During the last years, our group has developed chemical biology approaches to incorporate isotopically labelled amino acids into proteins in a site-specific manner, enabling the high-resolution NMR investigation of highly repetitive proteins, such as httex1.
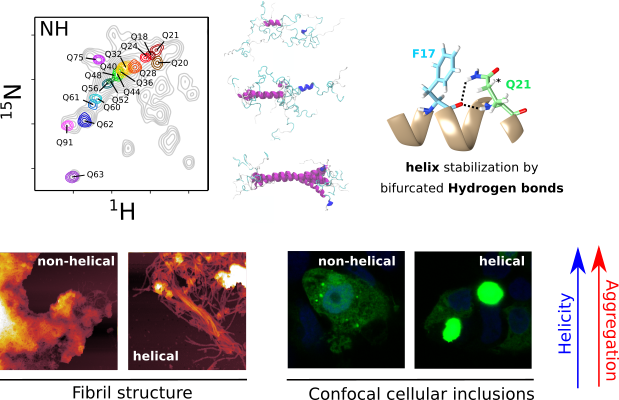
The systematic application of site-specific isotopic labelling has enabled the residue-specific NMR structural characterization of poly-Q tracts for pathogenic (Q46 and Q66) versions of httex1. These NMR data have been integrated with SAXS and computational approaches, to derive ensemble models and define the rules governing the structure of the poly-Q. Our analysis reveals that the poly-Q tract adopts long α-helical conformations propagated and stabilized by glutamine side chain to backbone hydrogen bonds, and highlights the relevance of the flanking regions in defining this structure. From a biomedical perspective, we show that α-helical stability is a stronger signature than the number of glutamines in defining aggregation kinetics, both in vitro and in cells, and the structure of the resulting fibrils. Our observations provide a structural perspective of the pathogenicity of expanded httex1 and pave the way to a deeper understanding of poly-Q-related diseases.
")
")